
فهرست مطالب
آلفا فولد، قهرمان علم زیستشناسی در کشف رازهای حیات
در دنیای پیچیده زیستشناسی، پروتئینها به عنوان مولکولهای کلیدی در عملکرد سلولها و ارگانیسمها شناخته میشوند. اما ساختار دقیق این پروتئینها چه تأثیری بر فعالیتهای بیولوژیکی آنها دارد. الفا فولد به عنوان یک فناوری پیشرفته، به ما این امکان را میدهد که به عمق ساختار پروتئینها نفوذ کنیم و رازهای پنهان آنها را کشف کنیم.
این ارائه به بررسی چگونگی عملکرد الفا فولد و تکنیکهای پیشرفته آن میپردازد که به دانشمندان کمک میکند تا ساختار سهبعدی پروتئینها را با دقت بینظیری پیشبینی کنند. همچنین، تأثیر این فناوری بر تحقیقات بیولوژیکی و پزشکی را تحلیل خواهیم کرد و نشان خواهیم داد که چگونه الفا فولد میتواند به توسعه درمانهای نوین و داروهای موثر در برابر بیماریهای مختلف کمک کند.
این سفر به اعماق پروتئینها، نه تنها به درک بهتر ما از حیات کمک میکند، بلکه میتواند درهای جدیدی به سوی درمانهای نوین و درک عمیقتری از بیماریها باز کند. توسعه یافته است که ساختار سه بعدی پروتئین را از روی توالی اسید آمینه آن پیش بینی می کند.
در CASP14، AlphaFold بهعنوان بهترین روش پیشبینی ساختار پروتئین با فاصلهای زیاد برگزیده شد و پیشبینیهایی با دقت بالا ارائه کرد. در حالی که این سیستم هنوز برخی محدودیتها را دارد، نتایج CASPنشان میدهد که AlphaFold پتانسیل فوری برای کمک به ما در درک ساختار پروتئینها و پیشبرد تحقیقات بیولوژیکی دارد.
الفا فولد یک سیستم هوش مصنوعی است که توسط گوگل دیپمایند توسعه یافته و ساختار سهبعدی یک پروتئین را از توالی اسیدهای آمینه آن پیشبینی میکند. این سیستم به طور مرتب دقتی رقابتی با آزمایشات را به دست میآورد.
گوگل دیپمایند و مؤسسه بیوانفورماتیک اروپا (EMBL-EBI) با هم همکاری کردهاند تاAlphaFold را ایجاد کنند و این پیشبینیها را بهطور رایگان در اختیار جامعه علمی قرار دهند. آخرین نسخه پایگاه داده شامل بیش از ۲۰۰ میلیون ورودی است که پوشش وسیعی از UniProt (مخزن استاندارد توالیهای پروتئینی و حاشیهنویسیها) را فراهم میکند. این پایگاه اطلاعات طبقه بندی شده ای برای پروتئوم انسان و پروتئوم ۴۷ ارگانیسم کلیدی دیگر که در تحقیقات و سلامت جهانی مهم هستند، ارائه میدهد.
پس از خواندن این مقاله پاسخ سوالات زیر را خواهید یافت:
- آلفا فولد چیست و چگونه کار میکند؟
- چه کاربردهایی برای آلفا فولد در علم زیستشناسی وجود دارد؟
- آلفا فولد چه مزایایی نسبت به روشهای سنتی دارد؟
- چالشها و محدودیتهای آلفا فولد چیست؟
- آینده آلفا فولد چگونه خواهد بود و چه تأثیری بر علم و فناوری خواهد گذاشت؟
تاریخچه پیشبینی ساختار پروتئین ها
پیشبینی ساختار پروتئینها یکی از چالشهای بزرگ در زیستشناسی مولکولی و بیوانفورماتیک است. این فرآیند از زمانهای قدیم آغاز شده و با پیشرفت تکنولوژی و الگوریتمها، تحولاتی را تجربه کرده است. در دهه 1970 با پیشرفت هایی در روش های تجربی و دستیابی به تکنیک هایی همچون کریستالوگرافی اشعه ایکس و NMR برای تعیین ساختار پروتئین ها به کارگرفته شد. در این میان نرم افزار های ابتدایی نیز برای پردازش کامپیوتری این پروتئین ها توسعه می یافتند ولی عمدتا به پیشرفت قابل توجهی منجر نمیشد.
در دهه 1980 با پیشرفت هایی در الگوریتم های بهینه سازی برای پیش بینی ساختار پروتئین ها بر اساس اصول ترمودینامیک حاصل شد. از طرفی پایگاه های داده ای برای پروتئین ها همچون PDB برای ذخیره و اشتراک گذاری اطلاعات ساختاری پروتئین ها ایجاد شدند.
در اوایل دهه 1990 کشف روش های محاسباتی مثل دینامیک مولکولی و شبیه سازی مونت کارلو با پیشرفت هایی در زمینه یادگیری ماشین (Machine learning) همراه بود. از سال 1994 با به وجود آمدن رقابتی به نام CASP (ارزیابی انتقادی پیش بینی ساختار پروتئین) پیشرفت چشمگیری در تلاش دانشمندان سراسر دنیا برای پیشبینی ساختار پروتئین ها با ابزار محاسباتی پدید آمد.
این روند تا سال 2016 ادامه داشت که با ظهور هوش مصنوعی یادگیری عمیق و با توسعه تیم DeepMind انقلابی در پیشبینی ساختار پروتئین ها ایجاد شد.
CASP (ارزیابی انتقادی پیشبینی ساختار) یک آزمایش جامع و بینالمللی است که به ارزیابی و مقایسه تکنیکهای پیشبینی ساختار پروتئین میپردازد. CASP یک رقابت عمومی است که به منظور تعیین وضعیت پیشرفته در مدلسازی ساختار پروتئین از توالی اسید آمینه سازماندهی شده توسط مرکز پیشبینی ساختار پروتئین انجام میشود. CASP یک مسابقه دوسالانه است که در آن شرکتکنندگان مدلهایی را ارائه میدهند که سعی در پیشبینی مجموعهای از پروتئینها دارند که ساختارهای تجربی آنها هنوز عمومی نشده است.
مدلهای ارائه شده با نتایج تجربی توسط گروه های مستقل مقایسه میشوند. این آزمایش به صورت کاملا محرمانه انجام میشود، به طوری که شرکتکنندگان به ساختارهای تجربی دسترسی ندارند و ارزیابان نیز نمیدانند که چه کسانی مدلها را ارائه کردهاند.
آلفافولد در رقابت های CASP
در دسامبر 2018، در رقابت CASP13 این آلفافولد بود که با یک جهشی قابل توجه نسبت به سال های پیش، برنده جایزه شد. این نخستین کاربرد موثر هوش مصنوعی در تعیین ساختار پروتئین ها بود. در نوامبر 2020، نسخه بهبود یافته از آلفافولد جایزه CASP14 را برد. به گفته یکی از بنیانگذاران CASP، جان مولت، آلفافولد در مقیاس 100 امتیازی دقت پیشبینی برای اهداف پروتئینی با درجه سختی متوسط، حدود 90 امتیاز کسب کرد. آلفافولد در سال 2021 به صورت متن باز منتشر شد و در CASP15 در سال 2022، در حالی که در رقابت های CASP شرکت نکرد، تقریباً تمام تیمهای با رتبه بالا از آلفافولد یا تغییرات آن استفاده کردند.
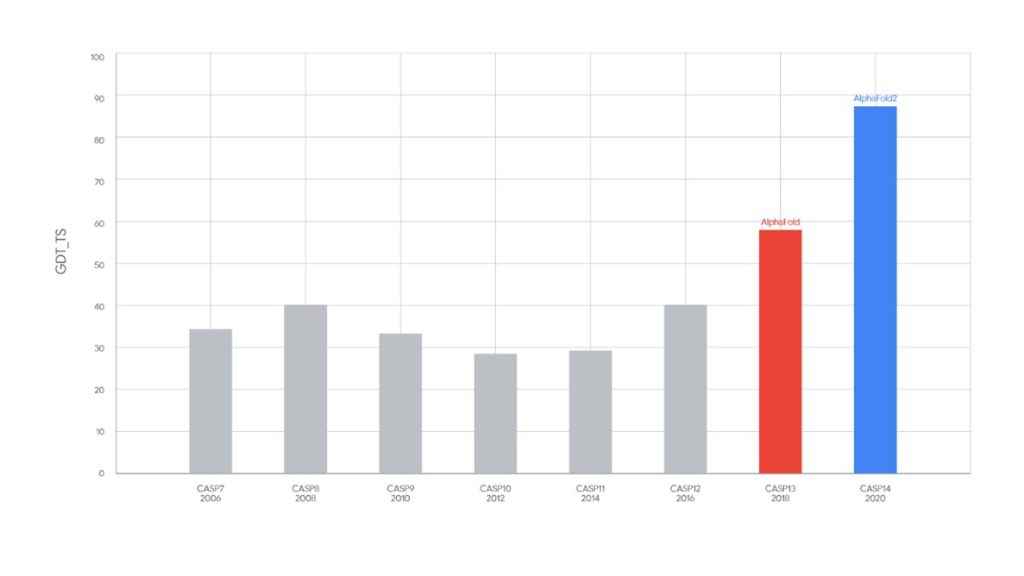
آلفافولد یک جهشی شگرف در پیشبینی ساختار پروتئین را نمایان میکند. آلفافولد قادر است دامنههای بیشتری را با دقت بالا نسبت به هر سیستم دیگری که در CASP13 شرکت کرده است، پیشبینی کند، بهویژه در محدوده امتیاز TM-score 0.6–0.7
آلفافولد با استفاده از تکنیکهای FM (تیم برنده CASP12) موفق به کسب z-score جمعی برابر با 52.8 شد که برای برنده قبلی 36.6 بود. علاوه بر این، با ترکیب تیم های FM و TBM/FM، AlphaFold امتیاز 68.3 را کسب کرد.
در مسابقه CASP14، آلفافولد2 به دستاوردی دست یافته است که میتوان آن را یک پیشرفت در مسئله تا شدن پروتئینها تلقی کرد. آلفافولد2 نتایج فوقالعادهای هم در مقایسه با سایر رقبا و همچنین در مقایسه با سایر برندگان سال های گذشته به دست آورد. راهحل دیپ مایند بهطور قابل توجهی از همه 145 راهحل دیگر شرکتکننده در مسابقه CASP14 پیشی گرفت و با نمره z مجموع 244.0217 به رتبه نخست دست یافت. این درحالی بود که نزدیکترین رقیب نمره z مجموع 92.1241 را کسب کرده بود. تقریباً دو سوم پیشبینیها از نظر کیفیت با ساختارهای تجربی قابل مقایسه بودند.
آلفافولد نتایجی خارج از مسابقه CASP نیز دارد. پیشبینیهای آلفافولد در برابر ساختار ORF3a اعتبارسنجی شدهاند، جایی که نتایج به دست آمده مشابه ساختارهایی بودهاند که بعداً بهطور تجربی تعیین شدهاند، با وجود طبیعت چالشبرانگیز آنها و داشتن توالیهای مرتبط بسیار کم.
اهمیت فولدینگ (تا خوردن) پروتئین
عملکرد بیولوژیکی یک پروتئین توسط ساختار سهبعدی آن تعیین میشود که در توالی اسید آمینهاش کدگذاری شده است. برای اینکه پروتئینها از نظر بیولوژیکی فعال باشند، باید ساختارهای سهبعدی خاصی را به خود بگیرند. تغییرات در ساختار پروتئین میتواند رفتار آن را به طور کامل تغییر دهد، به طوری که میتواند پروتئین را غیرفعال کرده و یا از بین ببرد. شناسایی و کنترل فولدینگ پروتئین بهطور قابلتوجهی بزرگترین چالش در زیستشناسی ساختاری است.
پروتئینهای دناتوره شده، که تقریباً تمام ساختار سهبعدی آنها مختل شده است، میتوانند از حالت نامنظم تصادفی خود به یک ساختار منحصر به فرد و مشخص دوباره فولد شوند، که در آن فعالیت بیولوژیکی تقریباً به طور کامل بازگردانی میشود. این نشان میدهد که آنها امکان بازسازی در محیط آزمایشگاهی را دارند و نشان میدهد که ساختار فولدینگ سهبعدی پروتئین در خود توالی کدگذاری شده و به فرآیند خارجی وابسته نیست.
پیچیدگی پیشبینی فولدینگ پروتئین
یکی از مشکلات در پیشبینی فولدینگ پروتئین ها این است که در حال حاضر راه آسانی برای محاسبه پتانسیل الکتروستاتیک دقیق، انرژیهای پیوندهایی که کمی کشیده شدهاند، یا در ساختارهای غیرایدهآل وجود ندارد. مگر اینکه به شبیهسازیهای مکانیک کوانتومی کند و بسیار پرهزینه متوسل شویم.
یکی دیگر از موانع مهم در پیشبینی فولدینگ پروتئین، وجود حالت های مختلف برای ساختار فضایی آن است. با فرض اینکه پیوندها دارای طول ثابت هستند، در یک پروتئین با طول متوسط 50 اسید آمینه (اندازه متوسط یک پروتئین انسانی 375 است)، هر اسید آمینه دارای دو پیوند چرخشی در ساختار اصلی است (زوایا سای و فی)، و تنها با در نظر گرفتن افزایشهای 10 درجه، به 10036 یا 1072 حالت ممکن میرسیم که عدد بسیار بزرگی است.
تکنیکهای پیشبینی ساختار
1) شبیهسازیهای فیزیکی
شبیهسازیهای فیزیکی، بر اساس شبیهسازی مکانیک کوانتومی، از نخستین تلاشها در پیشبینی فولدینگ پروتئینها به شمار میروند. ایده اصلی در اینجا اتکا به شبیهسازیهای دینامیک مولکولی برای به دست آوردن فرایند فولدینگ پروتئین است.
2) مونتاژ قطعه ای (Fragment Assembly)
مدلها از قطعههای کوتاه و متصل به هم از ستون فقرات (backbone) پروتئینهای با ساختار شناخته شده ساخته میشوند. انتخاب قطعهها معمولاً بر اساس شباهت توالی و همچنین پیشبینی ویژگیهای ساختاری محلی مانند ساختار ثانویه یا زوایای چرخشی backbone انجام میشود. تجمعهای چندگانهای از قطعهها تولید میشود و سپس با استفاده از یک معیار ارزش انرژی با دقت ارزیابی میشود. برای کاندیداهای با کمترین میزان انرژی (دارای پایداری بالا)، از یک معیار ارزش با دقت بالاتر و در نتیجه هزینه محاسباتی بیشتر استفاده میشود تا بهترین کاندید برای پیشبینی انتخاب شود. فرم تغییر یافته و پیشرفته تر این رویکرد به عنوان بهترین پیشبینیکنندهها در CASP9 و CASP10 شناخته شدند و در CASP11 نیز در میان دو ابزار برتر بودند.
3) ماشین لرنینگ
تکنیکهای ماشین لرنینگ تاریخچه طولانی در کاربردهای تحلیل ساختار پروتئین دارند. در ابتدا به عنوان اجزای یک جریان کاری برای پیشبینی ویژگیهای یکبعدی مانند زوایای چرخشی backbone یا ساختار ثانویه استفاده میشدند، اما اخیراً شاهد رونق این حوزه با رویکردهای انتها به انتها (end-to-end) بودهایم. آثاری همچون “یادگیری قابل تفکیک انتها به انتها از ساختار پروتئین و سپس معماریهای AlphaFold ایجاد شده توسط DeepMind قدرت واقعی پیشبینی یادگیری ماشین را نشان دادند. استراتژیهای موفق از پردازش تصویر و پردازش زبان طبیعی، مانند شبکههای عصبی کانولوشنی و مکانیزم توجه، در معماریهای طراحی شده برای پیشبینی چینش صحیح یک پروتئین گنجانده شدهاند.
آلفا فولد به عنوان ابزاری از خانواده هوش مصنوعی
همانطور که گفته شد، آلفافولد یک سیستم هوش مصنوعی است که برای حل مشکل تاخوردگی پروتئین طراحی شده است. این سیستم یک درکی نسبت به تکامل پروتئین ها دارد که شبکه عصبی کانولوشنی عمیق، به عنوان جزء اصلی آن عمل میکند. این سیستم توالیای را که برای آن فولدینگ مورد نظر است، همراه با همترازی توالیهای چندگانه (MSA) دریافت میکند. آلفافولد از الگوریتمی بهره میبرد که نشان میدهد زیرواحدها در تماس فضایی تمایل دارند الگوهای جهشهای همگرا را نشان دهند. (یعنی رفتاری را نشان میدهند که در طی روند تکامل در سایر پروتئین ها حفظ شده است.)
ساختار (معماری) آلفافولد
ساختار آلفافولد شامل دو بخش است:
1. شبکه عصبی کانولوشنی: این شبکه توالی پروتئین و ویژگیهای همراستای چندگانه را بهعنوان ورودی میگیرد و سطح پتانسیل خاص پروتئین را خروجی میدهد.
2. نزول گرادیان: در این مرحله، چندین اجرای نزول گرادیان برای پیدا کردن ساختاری که بهترین پایداری را برای انرژی پتانسیل پروتئین بهدست آورد، انجام میشود.
شبکه عصبی کانولوشنی عمیق
با استفاده از دیستوگرامها (هیستوگرامهای فاصله بین باقیماندهها)، میتوان مشکل پیدا کردن ساختار 3 بعدی پروتئین را مانند یک مشکل بینایی کامپیوتری تفسیر کرد. دیستوگرامها با استفاده از یک شبکه عمیق آمینواسید ها تولید میشوند. این شبکه فاصلههای بین اتمهای پروتئین و همچنین زوایای چرخشی بین آمینواسید ها را پیش بینی میکند.
نزول گرادیان بر روی پتانسیل خاص پروتئین
در این مرحله، یک میدان پتانسیل خاص پروتئین ساخته میشود که بر روی آن حداقل انرژی (بیشترین پایداری) با استفاده از نزول گرادیان محاسبه میشود. برای بهبود نتایج، یک الگوریتم ژنتیکی نیز برای پیدا کردن ساختاری با کمترین پتانسیل بهکار میرود.
آلفافولد2 نیز از یک جاسازی استفاده میکند که شامل دنبالههای کامل پیدا شده توسط MSA و الگوهای پتانسیل است، در حالی که راهحل قبلی تنها آمار MSA را دریافت میکرد. این به آلفافولد2 درک غنیتر و پیچیدهتری از فضای پروتئین میدهد.
جاسازی مفاهیم فیزیکی و هندسی در معماری به جای یک فرآیند جستجو انجام میشود. این بهطور قابل توجهی فضای جستجو را محدود کرده و عملکرد بهتری را با عدم کاوش یا حتی در نظر گرفتن پیکربندیهای فیزیکی غیرممکن فراهم میکند.
جایگزینی شبکه عصبی کانولوشن با یک معماری مبتنی بر توجه، جریان اطلاعات سفت و سخت از همسایگان محلی شبکههای کانولوشنی را با جریانی که بهطور دینامیک توسط شبکه کنترل میشود، جایگزین میکند.
استفاده از مدل بهصورت تکراری با استفاده از SE(3)-Transformer تکراری اجازه میدهد تا گرادیانها در سرتاسر معماری پراکنده شوند و قطع ارتباط بین پیشبینیهای فاصله جفتی و ساختار سه بعدی که در مدل اولیه آلفافولد مشاهده میشود، از بین برود.
کاربرد های آلفافولد
عملکرد فوقالعاده پیشبینی ساختار پروتئین توسط آلفافولد و انتشار بیش از ۲۰۰ میلیون ساختار پروتئین، در حال دگرگونی زیستشناسی ساختاری است و تأثیر عمیقی بر زمینههای زیستشناسی و پزشکی که به اطلاعات ساختاری پروتئین نیاز دارند، خواهد گذاشت. آلفافولد و ساختارهای پیشبینیشده آن به محققان این امکان را میدهد که فرصت بیشتری برای حل مسائلی که قبلاً به شدت چالشبرانگیز تلقی میشدند، داشته باشند. در ادامه، پیشرفتهای کاربردهای آلفافولد در زمینههای زیستشناسی و پزشکی را بررسی خواهیم کرد.
زیست شناسی ساختاری
بدون شک، زیستشناسی ساختاری بیشترین تأثیر را از آلفافولد دریافت کرده است. به جای اینکه بگوییم آلفافولد ممکن است زیستشناسان ساختاری را بیکار کند، ما ترجیح میدهیم به این دیدگاه اشاره کنیم که آلفافولد و ساختارهای پیشبینیشده آن، روشهای انجام زیستشناسی ساختاری را تغییر خواهند داد، از جمله کریستالوگرافی اشعه ایکس، میکروسکوپی الکترونی با دمای پایین (cryo-EM) و طیفسنجی NMR .
الف) استفاده به عنوان الگو: ساختارهای پیشبینیشده میتوانند به عنوان الگوهایی برای جایگزینی مولکولی در حل ساختارهای کریستالوگرافی اشعه ایکس مورد استفاده قرار گیرند، که نشان میدهد فازبندی سنتی سلنومتیونین تقریباً ضروری نیست.
ب) کمک به تعیین ساختار: این ساختارهای پیشبینیشده میتوانند در تعیین ساختار تجمعهای بزرگ پروتئینی با استفاده از cryo-EM مفید باشند، که معمولاً به ساختارهای پروتئینهای مؤلفه یا دامنههای آنها به عنوان نقطه شروع نیاز دارد.
ج) استفاده در :NMR همچنین میتوان از ساختارهای پیشبینیشده در استفاده از NMR برای حل ساختار پروتئین بهرهمند شد. معمولاً تعیین ساختار de novo دومین ها یا پروتئینها با استفاده از NMR که زمانبر است، ممکن است با ساختارهای AlphaFold جایگزین شود.
در حال حاضر، بسیاری از کاربردهای موفق در این زمینه وجود دارد. به عنوان مثال، Hu و همکاران از کریستالوگرافی اشعه ایکس و پیشبینی آلفافولد برای تعیین ساختار دامنه VP8 (VP8B) پروتئین راسی در روتاویروسهای گروه B استفاده کردند. در این مطالعه، نویسندگان پروتئین VP8B را بیان و خالصسازی کردند و سپس بلورهای این پروتئین را به دست آوردند و دادههای پراش اشعه ایکس را دریافت کردند. در فرآیند حل ساختار سهبعدی این پروتئین، به جای استفاده از روش سنتی فازبندی سلنومتیونین، از آلفافولد برای تولید یک مدل جستجوی مناسب برای جایگزینی مولکولی استفاده کردند. نتایج نشان داد که ساختار پیشبینیشده آلفافولد تقریباً بهطور کامل با چگالی پراش مطابقت دارد. علاوه بر شکل کلی، آلفافولد همچنین با دقت بالایی جهتگیری زنجیرههای جانبی را پیشبینی کرد که بسیار نزدیک به آنچه در آزمایشها تعیین شده بود، بود. نکته قابل توجه این است که آنها یک حالت جدید چینش را با آلفافولد پیدا کردند که هرگز در پروتئینهای همولوژی گزارش نشده بود.
Hutin و همکاران به تازگی ساختار هلیکاز DNA ویروس واکسینیا، هلیکاز-پرایمیز D5، را با استفاده از ترکیب cryo-EM و پیشبینی آلفافولد کشف کردند. ساختار بهدستآمده D5 یک هسته هلیکاز AAA+ را نشان میدهد که با دامنههای N- و C-terminal احاطه شده است. ساختار D5 پیشبینیشده توسط AF2 بهطور قابل توجهی به ساخت مدل کمک کرد. دامنه N-terminal که دارای وضوح ۳.۹ Å است، یک حلقه محکم و مشخص تشکیل میدهد، در حالی که وضوح به سمت C-terminus کاهش مییابد، که هنوز هم امکان تطابق ساختار پیشبینیشده را فراهم میکند. این ساختار محاسبات آلفافولد را برای تعداد زیادی از ساختارهای هلیکاز ویروسی مرتبط با D5 تأیید میکند.
Jin و همکاران ساختار کمپلکس سیگنال اینترلوکین (IL)−27 را با استفاده از cryo-EM و با کمک پیشبینی آلفافولد حل کردند، که از طریق آن یک مکانیسم جدید برای مونتاژ و فعالسازی کمپلکس شناسایی گیرنده IL-27 را منتشر کردند. Skalidis و همکاران از cryo-EM و آلفافولد برای توصیف ساختارهای متابولوندار یک پیشریبوزوم 60S، سنتاز اسید چرب و هستههای کمپلکس دی هیدروژناز پیرووات/اکسوگلوتارات E2 استفاده کردند. اگرچه بازسازیهای سهبعدی cryo-EM با وضوح بین ۳.۸۴ تا ۴.۵۲ Å از جمعآوری کمتر از ۳۰۰۰ میکروگراف از یک بخش سلولی منفرد حل شده بود، آلفافولد الگوهای پیوند هیدروژنی پلیپپتید را در این محدوده وضوح قابل تشخیص کرد. این نتایج یک رویکرد یکپارچه را پیشنهاد کردند که بهوسیله یادگیری ماشینی تقویت شده و امکان توصیف cryo-EM از عصارههای سلولی را فراهم میکند.
مطالعات مشابه دیگری نیز وجود دارد که در آنها آلفافولد به کمک تعیین ساختار استفاده میشود و برخی از این مطالعات آلفافولد و روشهای تجربی را ترکیب میکنند تا بررسی کنند آیا ساختار پروتئین به درستی حل شده است یا خیر، بهعنوان مثال، ترکیب آلفافولد با کریستالوگرافی اشعه ایکس، cryo-EM، NMR و چندین روش دیگر.
علاوه بر تعیین ساختار، پیشبینی آلفافولد حتی میتواند به طراحی ساختارهای بیان نیز اعمال شود. این قابلیتها به محققان این امکان را میدهد که بهتر تعیین کنند نقاط شروع و پایان یک دومین در توالی کجا واقع شده و از نواحی کمسازمان اجتناب کنند. نادیده گرفتن نواحی کمسازمان از توالیهای پروتئینی معمولاً به طراحی پروتئینهای نوترکیب برای تحقیقات مربوط به ساختارها کمک میکند.
کشف دارو با آلفافولد
کشف دارو یکی از زمینههای اصلی کاربرد است که به اطلاعات ساختاری پروتئین نیاز دارد. اگرچه سطح اعتماد به پیشبینیها متفاوت است، اما ساختارهای پیشبینیشده توسط آلفافولد میتوانند به طور قابل توجهی کشف داروهای مبتنی بر ساختار را ترویج دهند، به ویژه در برابر اهداف پروتئینی که اطلاعات ساختاری محدود یا هیچگونه اطلاعاتی ندارند. در حال حاضر، ساختارهای پروتئینی که در کشف داروهای مبتنی بر ساختار استفاده میشوند، عمدتاً از بانک داده پروتئینی (PDB) به دست میآیند. با این حال، تعداد ساختارهای پروتئینی در پایگاه داده PDB نسبتاً محدود است و از پاسخگویی به تقاضای فعلی برای کشف داروها فاصله دارد. انتظار میرود انتشار ساختارهای کل جهان پروتئین، پروژههای کشف داروی موجود و جدید را تسریع کند.
Zhang و همکاران به تازگی از برنامه داکینگ مولکولی Glide برای ارزیابی عملکرد غربالگری مجازی در برابر ۲۸ هدف دارویی رایج استفاده کردند که هرکدام دارای ساختار تجربی شناختهشده و یک ساختار آلفافولد هستند. ساختارهای آلفافولد در زمینه عامل غنیسازی، عملکرد قابل مقایسهای با ساختارهای تجربی نشان دادند. نتایج به وضوح نشان میدهند که ساختارهای آلفافولد میتوانند به طور کامل جایگزین ساختارهای تجربی در غربالگری مجازی شوند.
Ren و همکاران از AF2 در موتورهای کشف داروی مبتنی بر هوش مصنوعی خود استفاده کردند که شامل یک پلتفرم بیوانفورماتیک به نام پاندا اومیکس و یک پلتفرم شیمی سنتزی به نام Chemistry42 است. PandaOmics اهداف مورد نظر را فراهم میکند و Chemistry42 مسئول تولید مولکولها بر اساس ساختارهای پیشبینیشده آلفافولد است و سپس مولکولهای انتخابشده سنتز و در آزمایشهای بیولوژیکی آزمایش میشوند. از طریق این رویکرد، آنها یک ترکیب کوچک مؤثر برای CDK20 (کیناز وابسته به سایکلین20) با مقدار Kd برابر با ۸.۹ ± ۱.۶ میکرومول در ۳۰ روز از انتخاب هدف و پس از سنتز فقط ۷ ترکیب کشف کردند. این ترکیب در آن زمان اولین مولکول کوچک تارگت شده بر CDK20 بود و این کار اولین نمایش موفقیتآمیز آلفافولد در فرایند کشف داروی اولیه است.
Weng و همکاران از آلفافولد برای پیشبینی ساختار سهبعدی WSB1 (پروتئین WD-40) که یک هدف بالقوه جدید ضدسرطانی است و اطلاعات ساختاری سهبعدی آن در دسترس نیست، استفاده کردند. سپس ساختار پیشبینیشده با شبیهسازیهای دینامیک مولکولی بهینهسازی شد. ساختار سهبعدی بهینهشده WSB1 به عنوان ساختار گیرنده برای انجام داکینگ مولکولی به منظور غربالگری مهارکنندههای WSB1 استفاده شد. در نهایت، آنها تعدادی ترکیب فعال بالقوه به دست آوردند. در میان این ترکیبات، G490-0341 بهترین ساختار پایدار را نشان داد و شایسته تحقیقات و توسعه بیشتر بود.
Liang و همکاران JMJD8 را به عنوان یک آنکوژن جدید مرتبط با سرکوب ایمنی و ترمیم DNA از طریق تحلیل بیوانفورماتیک شناسایی کردند. سپس آنها از آلفافولد برای پیشبینی ساختار سهبعدی JMJD8 استفاده کرده و غربالگری مجازی برای بازیابی مهارکنندههای JMJD8 انجام دادند. Liu و همکاران یک روش کشف داروی چند هدفه پیشنهاد کردند و این روش را برای کشف داروهای هیپوترمی درمانی به کار بردند. در این مطالعه، آنها ابتدا ساختار تمام اهداف پروتئینی مرتبط را با استفاده از آلفافولد و RoseTTAFold پیشبینی کردند. پس از آن، آنها از داکینگ مولکولی برای تخمین تعامل بین پروتئینها و داروها استفاده کرده و داروهای تک یا ترکیبهای دارویی مناسب را تعیین کردند. با توجه به اختلاف وزنهای اهداف پروتئینی متفاوت، این رویکرد میتواند به طور مؤثری از مهار پروتئینهای مفید جلوگیری کند در حالی که پروتئینهای مضر را مهار میکند.
به جز مثالهای فوق، برخی تحقیقات نیز نشان دادند که کیفیت زنجیره جانبی مدلشده توسط آلفافولد برای کشف دارو کافی نیست و برخی مطالعات اخیر نیز نشان دادند که آزمون داکینگ مبتنی بر ساختارهای پیشبینیشده آلفافولد عملکرد ضعیفی دارد.
طراحی پروتئین با آلفافولد
طراحی پروتئین به معنای ایجاد پروتئینهای جدید با ساختارها و عملکردهای مورد نظر است. طراحی پروتئین به صورت -de novo یک هدف بنیادی دیرینه در زیستشناسی سنتزی است. این یک وظیفه پیچیده و چالشبرانگیز است که عمدتاً به دلیل دشواری در پیشبینی قابل اعتماد ساختارهای سهبعدی پروتئین از توالیهای اسید آمینه محدود شده است. آلفافولد و سایر الگوریتمهای یادگیری ماشین (مانند RoseTTAFold و مدلهای زبانی اخیر) احتمالاً این مانع را برطرف میکنند. به هیچ وجه نمیتوان اغراق کرد که با پیشبینی آلفافولد ، ما وارد یک عصر جدید در طراحی پروتئین خواهیم شد. برخی از مثالهای معمول طراحی پروتئین با استفاده از آلفافولد به شرح زیر است:
Jendrusch و همکاران یک چارچوب محاسباتی برای طراحی پروتئین به صورت de novo توسعه دادند که آلفافولد را به عنوان یک پیشبینی کننده در یک فرآیند طراحی قابل بهینهسازی قرار میدهد. این یک چارچوب قابل تنظیم برای طراحی پروتئین از طریق بهینهسازی توالی با استفاده از الگوریتمهای تکاملی است. این کار به مطالعات قبلی در زمینه طراحی پروتئین با بهرهگیری از پیشبینیکنندههای ساختاری گسترش مییابد. تمامیت ساختارهای پیشبینیشده با استفاده از روشهای استاندارد تجزیه و تحلیل ساختار پروتئین و شبیهسازیهای دینامیک مولکولی دقیق تأیید و اعتبارسنجی شده است. آنها همچنین یک کاربرد بالقوه از روش خود را در طراحی مونومرها، دیمرها و الیگومرهای پروتئینی جدید، و همچنین بایندرهای پروتئینی برای پروتئینهای هدف و پروتئینهایی که در هنگام تشکیل کمپلکس تغییر شکل میدهند، نشان دادند.
پیشبینی تارگت مولکول با آلفافولد
پیشبینی تارگت، شامل شناسایی هدف اصلی و غیرهدف، نه تنها برای درک فرآیندهای فیزیولوژیکی و پاتولوژیکی مهم است، بلکه برای شناسایی اهداف دارویی جدید و ارزیابی انتخابپذیری داروها نیز ضروری است. روشهای تجربی برای شناسایی تارگت، مانند روشهای مختلف مبتنی بر نمایه سازی پروتئین بر اساس فعالیت (ABPP)، اغلب هزینهبر و زمانبر هستند. پیشبینی تارگت با کمک کامپیوتر میتواند دامنه شناسایی هدف را محدود کند که معمولاً بر اساس داکینگ پروتئین-لیگاند، که معمولاً به آن داکینگ معکوس گفته میشود، انجام میشود. پیشتر، داکینگ معکوس با چالشی از کمبود ساختارهای سهبعدی تمام پروتئینهای ممکن مواجه بود. ساختارهای آلفافولد فرصتی بیسابقه برای توسعه روشهای پیشبینی تارگت قابل اجرا فراهم میکنند.
Wang و همکاران از ساختارهای آلفافولد برای ساخت اولین پایگاه داده برای تمام پروتئینهای موجود در پروتئوم انسانی استفاده کردند که به آن پایگاه داده CavitySpace میگویند. CavitySpace میتواند برای شناسایی اهداف جدید برای داروهای شناختهشده در تحقیقات بازپیکربندی دارو یا عوارض جانبی استفاده شود. این پایگاه داده به راحتی میتواند برای پیشبینی هدف از طریق داکینگ معکوس مورد استفاده قرار گیرد. روند ساخت این پایگاه داده به شرح زیر است: آنها ۲۳,۳۹۱ پروتئین انسانی را از پایگاه داده ساختار پروتئین آلفافولد و ۶,۹۵۶ پروتئین مرجع انسانی را از PDB جمعآوری کردند. سپس، آنها از CAVITY، ابزاری که توسط همان گروه تحقیقاتی برای شناسایی تمام حفرههای ممکن بر روی سطوح پروتئینی توسعه یافته است، برای شناسایی تمام حفرههای بالقوه بر روی سطوح پروتئینها استفاده کردند. پایگاه داده CavitySpace به صورت رایگان در http://www.pkumdl.cn:8000/cavityspace در دسترس است. همچنین مطالعات دیگری وجود دارد که آلفافولد را برای پیشبینی هدف به کار بردهاند.
پیشبینی عملکرد پروتئین با آلفافولد
در حال حاضر، هنوز بسیاری از پروتئینها وجود دارند که عملکرد آنها ناشناخته یا به خوبی تعیین نشده است. از آنجا که ساختارهای سهبعدی پروتئینها به طور کامل عملکرد آنها را تعیین میکنند، این ویژگی میتواند برای ایجاد مدلهای پیشبینی عملکرد پروتئین مبتنی بر دادهها مورد استفاده قرار گیرد. با این حال، تعداد ناکافی ساختارهای پروتئینی موجود، عملکرد این مدلها را به شدت محدود میکند. ساختارهای پیشبینیشده توسط آلفافولد یک راهحل امیدوارکننده برای این مشکل ارائه میدهند و انتظار میرود که با افزایش تعداد نمونههای آموزشی، عملکرد این مدلها را بهبود ببخشند.
Ma و همکاران اخیراً یک مطالعه جامع انجام دادند تا بررسی کنند آیا ساختارهای پیشبینیشده توسط آلفافولد میتوانند عملکرد پیشبینی عملکرد پروتئین را بهبود بخشند. در این مطالعه، آنها یک رویکرد پیشرفته مبتنی بر ساختار برای پیشبینی عملکرد پروتئین پیشنهاد کردند و یک مجموعه داده مرجع جدید ساختند. سپس آنها ارزیابی کردند که آیا عملکرد مدل پیشبینی عملکرد پروتئین میتواند با اضافه کردن ساختارهای پروتئینی پیشبینیشده توسط آلفافولد به مجموعه داده آموزشی بهبود یابد. آنها همچنین تفاوتهای عملکرد بین دو مدل را که بهطور جداگانه با ساختارهای پیشبینیشده توسط آلفافولد و فقط با ساختارهای واقعی پروتئین آموزش داده شده بودند، مقایسه کردند. نتایج آنها نشان داد که مدلهای پیشبینی عملکرد پروتئین مبتنی بر ساختار میتوانند از دادههای بیوانفورماتیکی که شامل ساختارهای پیشبینیشده توسط آلفافولد هستند، بهرهمند شوند. حتی مدل آموزشدیده تنها با استفاده از ساختارهای پیشبینیشده توسط آلفافولد2 عملکرد قابل مقایسهای با مدل مبتنی بر ساختارهای واقعی پروتئین که از طریق آزمایشات حل شدهاند، داشت. این نشان میدهد که ساختارهای پیشبینیشده توسط آلفافولد تقریباً به طور یکسان در پیشبینی عملکرد پروتئین مؤثر بودند.
نمایشهای ویژگی ساختاری قابل تفسیر و فشرده برای پیشبینی دقیق خواص و عملکرد پروتئینها مهم هستند. در یک مطالعه اخیر، Rappoport و Jinich نمایشهای ویژگی سهبعدی ساختارهای پروتئینی را با استفاده از منحنیهای پرکننده فضا ساختند و ارزیابی کردند که در آن ساختارهای پروتئینی پیشبینیشده توسط آلفافولد استفاده شدند. در این مطالعه، دو پیشبینی زیرلایه آنزیمی به عنوان مطالعات موردی استفاده شد: متیلانتقالدهندههای وابسته به S-ادنوزین متیونین (SAM-MTases) و دهیدروژناز/کاهشدهندههای زنجیره کوتاه (SDRs). همانطور که نتایج آنها نشان میدهد، عملکرد آنزیمی میتوانست از نمایشهای ویژگی بر اساس ساختارهای سهبعدی SAM-MTAses و SDRs با دقت خوبی پیشبینی شود.
با جستجوی پروتئینهایی که حاوی دامنه Zα (دامنه پروتئینی Z-DNA/Z-RNA که بهطور تجربی تأیید شده است) از پایگاه داده ساختار پیشبینیشده آلفافولد2 هستند، Bartas و همکاران ۱۸۵ پروتئین با یک دامنه Zα احتمالی شناسایی کردند که ممکن است به Z-DNA/Z-RNA متصل شوند و نقش مهمی در فرآیندهای سلولی مختلف ایفا کنند. همچنین تحقیقات جالب دیگری وجود دارد که آلفافولد را در پیشبینی عملکرد پروتئین دخیل میکند.
بررسی تعاملات پروتئین-پروتئین با آلفافولد
تعامل پروتئین-پروتئین (PPI) به فرآیندی اشاره دارد که در آن دو یا چند مولکول پروتئینی از طریق پیوندهای غیرکووالانسی یک کمپلکس پروتئینی تشکیل میدهند. اکثر پروتئینها برای انجام عملکردهای خود نیاز دارند که پروتئینهای دیگر را از طریق پروتئین-پروتئین اینترکشن (PPI) جذب کنند. درک ساختار پروتئینهای در حال تعامل، یک گام اساسی در راستای کشف عملکرد و مکانیزم پروتئینها است. با این حال، ابزارهای محاسباتی کافی برای تولید ساختارهای دقیق کمپلکسهای پروتئینی وجود ندارد. ظهور آلفافولد میتواند به شدت به این حوزه کمک کند.
Evans و همکاران آلفافولد را برای پیشبینی کمپلکسهای چند زنجیرهای گسترش دادند و این سیستم AlphaFold-Multimer نامیده شد. در یک مجموعه داده مرجع از ۱۷ پروتئین هترودیمر بدون الگو، آنها حداقل دقت متوسطی را در ۱۴ هدف و دقت بالایی را در ۶ هدف به دست آوردند. آنها همچنین ساختارهایی را برای یک مجموعه داده بزرگ شامل بیش از ۴,۰۰۰ کمپلکس پروتئینی اخیر پیشبینی کردند و از این میان، تمام رابطهای غیر تکراری با هویت الگوی پایین را از این کمپلکسهای پروتئینی امتیازدهی کردند. برای رابطهای هترومر، آنها موفق به پیشبینی رابط در ۶۷٪ موارد شدند و ۲۳٪ از موارد با دقت بالا پیشبینی شدند. برای رابطهای هومومر نیز در ۶۹٪ موارد رابط را بهطور مؤثر پیشبینی کردند و در ۳۴٪ از موارد پیشبینیهای با دقت بالایی تولید کردند. تمام این نتایج عملکرد برتری را در مقایسه با رویکردهای موجود نشان دادند. AlphaFold2-multimers اکنون برای پیشبینی ساختارهای کمپلکس پروتئین-پروتئین استفاده میشود. بهعنوان مثال، Gómez-Marín و همکاران از AlphaFold-multimer برای پیشبینی مدلهای کمپلکس PHF14-HMG20A استفاده کردند. Ivanov و همکاران نیز از AlphaFold-multimer برای پیشبینی ساختار هومودیمر CYP102A1 استفاده کردند.
بهتازگی، Bryant و همکاران از آلفافولد برای پیشبینی کمپلکسهای پروتئینی هترودیمر استفاده کردند. در این کار، آنها اثر داکینگ را با استفاده از خط لوله آلفافولد2 ترکیبشده با MSAs ورودی مختلف بررسی کردند تا رابطه بین کیفیت مدل خروجی و این ورودیها را مطالعه کنند. با امتیازدهی به مدلهای PPI متعدد با یک امتیاز DockQ پیشبینیشده (pDockQ)، آنها توانستند مدلهای نادرست را با اطمینان بالا از یکدیگر تشخیص دهند (pDockQ ≥ 0.23). آنها نتیجهگیری کردند که داکینگ مبتنی بر آلفافولد عملکرد بهتری نسبت به یک روش داکینگ دیگر دارد.
Yin و همکاران عملکرد آلفافولد را در پیشبینی ساختارهای کمپلکسهای پروتئینی از توالیهای آمینو اسید بررسی کردند. آنها از ۱۵۲ کمپلکس پروتئینی هترودیمر متنوع برای تشکیل یک مجموعه داده آزمایشی مرجع استفاده کردند. در این آزمایش، ۴۳٪ از مواردی که مدلهای نزدیک به حالت طبیعی داشتند بهعنوان نتایج پیشبینی برتر توسط آلفافولد تولید شدند که بهطور قابل توجهی عملکرد روش داکینگ پروتئین–پروتئین بدون اتصال (۹٪) را بهتر کردند. برای بررسی اثر AlphaFold_Multimer در پیشبینی تعامل آنتیبود–آنتیژن، نویسندگان از مجموعهای از ساختارهای آنتیبود–آنتیژن استفاده کردند که بهتازگی منتشر شده بودند. نتایج آزمایش تأیید کردند که نرخ موفقیت پایینی برای مدلسازی کمپلکسهای آنتیبود–آنتیژن وجود دارد. آنها همچنین مشاهده کردند که از طریق الگوریتم، کمپلکسهای گیرنده T cell–آنتیژن نیز بهطور دقیق مدلسازی نشدهاند. این یافتهها نشان میدهند که آلفافولد در مدیریت شناسایی ایمنی تطبیقی با چالشهایی مواجه است. Gao و همکاران یک سیستم مبتنی بر آلفافولد به نام AlphaFold Complex توسعه دادند که میتواند تعاملات فیزیکی مستقیم را در پروتئینهای چند زنجیرهای پیشبینی کند. برخلاف رویکردهای معمول، جفتهای MSAs برای AlphaFold Complex ضروری نیستند. این سیستم بهطور قابل توجهی از AlphaFold-Multimer بهبود یافته و دقت بالاتری را در مقایسه با برخی از روشهای داکینگ پروتئین–پروتئین پیچیده به دست میآورد. علاوه بر این، نویسندگان معیارهایی را برای پیشبینی تعاملات مستقیم پروتئین–پروتئین بین جفتهای دلخواه پروتئین معرفی کردند و AlphaFold Complex را بر روی پروتئوم E. coli و همچنین برخی مجموعههای آزمایشی چالشبرانگیز اعتبارسنجی کردند.
علاوه بر PPI، آلفافولد همچنین میتواند در پیشبینی تعاملات پپتید–پروتئین استفاده شود. بهعنوان مثال، Tsaban و همکاران یک استراتژی مبتنی بر آلفافولد برای مدلسازی کمپلکس پپتید–پروتئین پیشنهاد کردند که نیازی به اطلاعات MSA برای شریک پپتید نداشت. به این ترتیب، تغییرات کنفورماسیونی ناشی از اتصال گیرنده قابل مدیریت بودند. نتایج نشان میدهد که آلفافولد میتواند انتظار داشته باشد که بینش ساختاری در مورد دامنه وسیعی از کمپلکسهای پپتید–پروتئین ارائه دهد.
بررسی مکانیسم عمل بیولوژیکی اغلب پیچیده است و چالشی باقی میماند. مطالعات مکانیسم عمل بیولوژیکی شامل جنبههای مختلفی از جمله نحوه تعامل دارو با هدف، مکانیزم کاتالیز آنزیمهای بیولوژیکی و غیره میباشد.
روشهای داکینگ مولکولی در شبیهسازیهای کامپیوتری بهطور گستردهای برای پیشبینی تعامل دارو و هدف استفاده شدهاند. با این حال، این نوع روشها به شدت به ساختارهای پروتئینی موجود وابسته هستند. آلفافولد یک رویکرد جایگزین برای بهدست آوردن ساختارهای دقیق پروتئین فراهم میکند. Wong و همکاران داکینگ مولکولی را با AF2 برای پیشبینی تعاملات پروتئین-لیگاند ترکیب کردند. آنها با موفقیت تعاملات بین ۲۹۶ پروتئین که شامل پروتئوم اساسی Escherichia coli بود و ۲۱۸ ترکیب آنتیبیوتیکی فعال و ۱۰۰ ترکیب غیر فعال را پیشبینی کردند. آنها فعالیت آنزیمی ۱۲ پروتئین اساسی را که با هر ترکیب آنتیبیوتیکی درمان شده بودند، اندازهگیری کردند تا عملکرد مدل را ارزیابی کنند. این تحقیق نشان میدهد که رویکردهای پیشرفته در مدلسازی تعاملات پروتئین-لیگاند، بهویژه با استفاده از روشهای مبتنی بر یادگیری ماشین، برای بهرهبرداری بهتر از آلفافولد در مطالعات مکانیسم عمل و کشف داروها نیاز است.
اکسیداسیون لاکتات با NAD+ بهعنوان پذیرنده الکترون یک واکنش بسیار انرژیبر است. برخی از باکتریهای بیهوازی با استفاده از انشعاب/ادغام الکترون (FBEB/FBEC) مبتنی بر فلافین، که شامل لاکتات دهیدروژناز (Ldh) به همراه EtfA و EtfB است، که پروتئینهای انتقالدهنده الکترون هستند، بر این مانع انرژی غلبه میکنند. با این حال، مکانیزم عمل بهخوبی درک نشده است. در یک مطالعه اخیر، Kayastha و همکاران از محاسبات آلفافولد استفاده کردند و یک حالت جدید B (متصل به انشعاب) قابل قبول بهدست آوردند که اجازه میدهد الکترون بین FAD های شاتل و پایه EtfAB منتقل شود. بر اساس یافتهها، آنها یک مکانیزم کاتالیز یکپارچه برای فرآیند FBEC ارائه دادند.
Liang و همکاران تعیینکنندههای زیرلایه و مکانیزم شناسایی سپاراز، که یک پروتئاز سیستئینی غولپیکر است، را بررسی کردند. در مخمر جوانهزنی، آنها یک موتیف محافظتشده در پاییندست محل برش شناسایی کردند. با استفاده از آلفافولد و شبیهسازیهای دینامیک مولکولی، آنها کشف کردند که در یک شکاف محافظتشده نزدیک به شیار اتصالدهنده مهارکننده سپاراز، این موتیف توسط سپاراز شناسایی میشود. این اتصال بهطور متقابل انحصاری است و نیاز به تغییر شکل سپاراز دارد. تحقیقات آنها میتواند به دانشمندان کمک کند تا درک عمیقتری از مکانیزم شناسایی زیرلایه و فعالسازی سپاراز بهدست آورند.
Lorenz و همکاران از آلفافولد برای پیشبینی ساختار دامنههای KRAB انتخابشده استفاده کردند. آنها یک بدنه L شکل محافظتشده از دو α-هلیکس را در تمام دامنههای KRAB کشف کردند. این ساختار به یک آرایش فضایی معمولی بهویژه برای mKRABAB پس از جایگزینی اسید آمینه و همراه با یک هلیکس سوم ارائه شده توسط mKRAB-B تغییر یافت. این موضوع بینشهای بنیادی درباره چگونگی تشکیل کمپلکس KRAB با TRIM28 ارائه میدهد. McMullen و همکاران از مخمر دریافتند که EKP-GCSF و GCSF بهطور مشابه به گیرنده GCSF-R متصل میشوند. بهطور مشابه، برای مطالعه اثرات ساختاری EKP بر GCSF، آنها مدلسازی محاسباتی را با استفاده از آلفافولد در ترکیب با شبیهسازیهای دینامیک مولکولی انجام دادند. مدلسازی محاسباتی نشان میدهد که EKP رفتار ساختاری GCSF را تغییر نمیدهد، که نشان میدهد EKP مانع اتصال گیرنده نمیشود. علاوه بر این، شکل اولیه EKP-GCSF از AlphaFold نشان میدهد که EKP که در اطراف GCSF قرار دارد ممکن است شواهدی از محافظت حرارتی EKP بر GCSF ارائه دهد.
کاربردهای دیگر آلفافولد
علاوه بر حوزههای کاربردی ذکر شده در بالا، پیشبینی آلفافولد همچنین میتواند در برخی دیگر از زمینهها مانند تکامل پروتئین، مطالعات درمان بیماریهای نادر، اثرات جهش بر درمان، طراحی واکسن و غیره مورد استفاده قرار گیرد. بهعنوان مثال، Tang و همکاران رابطه بین تکامل ارگانیسم و تکامل پروتئین را بر اساس ساختارهای پروتئوم ۴۸ ارگانیسم پیشبینیشده توسط آلفافولد بررسی کردند. آنها برخی پدیدههای جالب را کشف کردند، از جمله: (۱) پروتئینهای سازنده ارگانیسمهای با پیچیدگی بالاتر دارای شعاع چرخش بزرگتر، نسبتهای مارپیچی بالاتر و لرزشهای کندتر هستند، و (۲) درجه بالاتر تخصص عملکردی پروتئینها با درجه بالاتر پیچیدگی ارگانیسمها مرتبط است. این تحقیق دیدگاههای جدیدی درباره چگونگی افزایش تنوع عملکردی پروتئینها و کاهش ابعاد منیفولد دینامیک پروتئینها در فرآیند تکامل ارائه میدهد. Sebastiano و همکاران دریافتند که ساختارهای پروتئینی پیشبینیشده توسط آلفافولد پتانسیل کمک به مطالعات درمان بیماریهای نادر را دارند. در این تحقیق، نویسندگان بر روی Alsin، پروتئینی که مسئول بیماریهای نادر نورون حرکتی است، تمرکز کردند. با استفاده از ساختارهای پروتئینی پیشبینیشده توسط AF2، آنها پروفایل انعطافپذیری Alsin و جهشهای آن و مدلهای دیمری/تترامری Alsin مسئول عمل فیزیولوژیکیاش را ارزیابی کردند. آنها نتیجهگیری کردند که تلاشهای کشف دارو برای بیماریهای مرتبط با Alsin باید ادامه یابد. Yang و همکاران از آلفافولد برای پیشبینی ساختارهای پروتئینهای S، N و M واریانت Omicron SARS-CoV-2 استفاده کردند. آنها بهطور دقیق تحلیل کردند که چگونه پروتئین S و اجزای آن، S1 RBD و NTD تحت تأثیر جهشها قرار گرفتهاند و همچنین چگونه واکسنها و درمانهای فعلی SARS-CoV-2 تحت تأثیر این جهشها قرار خواهند گرفت. Zeng و همکاران از آلفافولد برای طراحی یک واکسن ساقه هماگلوتینین به نام “B60-Stem-8070” استفاده کردند. این واکسن عملکرد بهتری نسبت به آنتیژن ساقه هماگلوتینینین اصلی نشان داد.
آلفافولد برنده جایزه نوبل شیمی 2024
در سال گذشته جایزه نوبل شیمی 2024 به دیوید بیکر، جان جامپر و دمیس هسابیس برای ارائه آلفافولد تعلق گرفت.
دیوید بیکر (David Baker) متولد 6 اکتبر 1962 (14 مهر 1341) در آمریکا، بیوشیمیست و زیستشناس محاسباتی است که روشهایی را برای طراحی پروتئینها و پیشبینی ساختارهای سهبعدی آنها راهاندازی کرده است. او استاد بیوشیمی هنریتا و اوبری دیویس، محقق مؤسسه پزشکی هاوارد و استادیار علوم ژنوم، مهندسی ژنتیک، مهندسی شیمی و علوم کامپیوتر در دانشگاه واشنگتن است. بیکر و همکارانش با توسعه برنامههای هوش مصنوعی، موفق به حل مسائل مربوط به پیشبینی ساختار پروتئین شدند. او همچنین در سال 2022 جایزه وایلی را دریافت کرد. گروه تحقیقاتی بیکر یکی پس از دیگری پروتئینهای مصنوعی تولید کرده است. از بعضی از این پروتئینها میتوان بهعنوان دارو، واکسن و مواد نانو و حسگرهای کوچک استفاده کرد.
جان مایکل جامپر (John M. Jumper) دانشمند آمریکایی شرکت هوش مصنوعی دیپمایند است. جامپر و همکارانش، آلفا فولد را طراحی کردند که یک مدل هوش مصنوعی برای پیشبینی ساختارهای پروتئینی از توالی اسیدهای آمینه آنها با دقت بالا است. جامپر در دانشگاه شیکاگو تحصیل کرده است.
دمیس هسابیس (Demis Hassabis) متولد 27 ژوئیه 1976 (5 مرداد 1355) دانشمند علوم رایانه، محقق هوش مصنوعی و کارآفرین بریتانیایی است. او در ابتدای کار خود برنامهنویس و طراح بازیهای ویدیویی هوش مصنوعی بود. وی مدیر اجرایی و یکی از بنیانگذاران شرکت هوش مصنوعی دیپمایند و Isomorphic Labs و نیز مشاور هوش مصنوعی دولت انگلیس است.

چالش ها و محدودیت های آلفافولد
با اینکه آلفافولد یک انقلابی در زیستشناسی ساختاری است، اما در حال حاضر تا حد زیادی بر روی ساختارهای پروتئینی موجود در بانک داده پروتئین (Protein Data Bank) استوار است که در که به وسیله ی کریستالوگرافی اشعه ایکس به دست آمده اند. بنابراین، بیشتر بهعنوان یک پیشبینیکننده وضعیت ساختاری بر اساس شرایط تجربی شناخته می شود که از پیشبینی برخی از استثناهایی که ممکن است به وقوع بپیوندد ناتوان است.به علاوه برخی محدودیتهای ذاتی در روشها و تکنیکها، کاربردهای پیشبینی آلفافولد را در بسیاری از جنبهها محدود میکند که بهطور خلاصه به شرح زیر است:
1) دینامیک پروتئین
دینامیک پروتئین یک حوزه تحقیقاتی بسیار مهم است. ساختار پروتئین پیشبینیشده توسط آلفافولد یک حالت ایستا است. با این حال، پروتئینها بسیار پویا هستند و دارای چندین حالت مختلف میباشند. بسیاری از پروتئینهای فیزیولوژیکی و پاتولوژیکی مهم (مانند پروتئینهای کانال یونی) تغییرات کنفورماسیونی بسیار ظریفی در حالتهای فعال مختلف دارند و همچنین به دلیل ترکیب با پروتئینهای مختلف در داخل و خارج از سلول، اشکال فضایی متغیری را نشان میدهند. اما آلفافولد معمولاً تنها یک شکل از آن پروتئین را ارائه میدهد که دشوار است تنوع کنفورماسیونی پروتئینها را پوشش دهد. با این حال، این به این معنا نیست که نمیتوان دینامیک پروتئین را با آلفافولد مشاهده کرد. بر اساس چندین مطالعه اخیر، آلفافولد هنوز هم میتواند برای برخی تحلیلهای دینامیک پروتئین مورد استفاده قرار گیرد. به عنوان مثال، اخیراً به کمک آلفافولد روش هایی برای نمایش کنفورماسیون های جایگزین ترانسپورترهای توپولوژیکی متنوع و همچنین گیرندههای انتقال سیگنال G-protein-coupled را ارائه دادند. مطالعات حاکی از آن است که آلفافولد در شناسایی دقیق کنفورماسیونی پروتئین های بزرگ با چندین دمین (مثل یک گیرنده تراغشایی با دامنه بزرگ خارجسلولی)، محدودیت هایی دارد که اشاره به لزوم طراحی روشهای جدید یادگیری عمیق برای پیشبینی مجموعهای از حالات بیوفیزیکی مرتبط دارد.
2) ساختارهای نواحی بینظم پروتئینها
آلفافولد در پیشبینی ساختار پروتئینها در مواردی که توالیهای کمتری برای مقایسه همپوشانی در دسترس باشد و نواحی که از نظم طبیعی درون مولکولی پیروی نمیکنند، مانند لوپها، عملکرد خوبی ندارد. ساختارهای لوپ در حالت جامد نسبتاً پایدار هستند، اما در محلولها بسیار ناپایدارند. با وجود تمام روشهای جدید هنوز هم آلفافولد برای پیشبینی مورفولوژی، دینامیک و تعاملات نواحی بینظم پروتئینها و به ویژه در حالت محلول با چالش هایی روبه رو است.
3) ساختارهای پروتئینها در ترکیب با مولکولهای کوچک یا پروتئینهای دیگر
لیگاندهای مولکولهای کوچک یا پروتئینها ممکن است باعث تغییر شکل یک پروتئین شوند. مثال بارز این تغییرات، برخی مولکولهای کوچک یا پپتیدهایی هستند که به محلی از پروتئین آنزیم متصل میشوند که متفاوت از محل اتصال لیگاند اندوژن است تا تغییرات کنفورماسیونی ایجاد کند و در نتیجه فعالیت آنزیم را تغییر دهد. علاوه بر این مولکول ها، بسیاری از لیگاندها که به محل لیگاند متصل میشوند نیز میتوانند تغییرات کنفورماسیونی ایجاد کنند. آلفافولد برای تعیین اینکه پروتئینها چگونه در حضور لیگاندها یا پروتئینهای دیگر تغییر شکل میدهند، طراحی نشده است.
4) ساختارهای پروتئینها با جهشهای نقطهای
جهشهای نقطهای بهطور مکرر در پروتئینها، بهویژه در حالت پاتولوژیک، مشاهده میشوند. درک اثر جهشهای نادرست بر ساختار پروتئین ممکن است به کشف مکانیزمهای بیولوژیکی یا پاتولوژیکی آنها کمک کند. حتی اگر آلفافولد بتواند ساختارهای نوع وحشی (WT) را پیشبینی کند، احتمالاً در پیشبینی اثر جهشهای نادرست بر ساختارهای سهبعدی پروتئینها عملکرد ضعیفی دارد. اگرچه تحقیقات نشان میدهند که آلفافولد میتواند اثر فنوتیپی جهشهای نادرست را پیشبینی کند، اما همچنین مشاهده شده است که عملکرد پیشبینی جهشهای نادرست در سایر مطالعات دقیق نیست و فقط ارتباطات ضعیف یا عدم ارتباط بین معیارهای خروجی آلفافولد و تغییرات در پایداری یا عملکرد پروتئین وجود دارد.
5) ساختارهای پروتئینها با تغییرات پس از ترجمه
تغییرات پس از ترجمه، مانند فسفوریلاسیون، متیلاسیون، استیلاسیون و گلیکوزیلاسیون، در پروتئینها رایج هستند. این تغییرات پس از ترجمه ممکن است منجر به تغییرات کنفورماسیونی در ساختارهای پروتئینی شوند. به عنوان مثال، فسفوریلاسیون کینازهای غیرفعال در لوپ فعال آنها معمولاً منجر به تغییرات کنفورماسیونی بزرگ میشود و در نهایت کینازها را فعال میکند. با این حال، آلفافولد فقط میتواند ساختار پروتئینها را بر اساس توالی آمینو اسید آنها پیشبینی کند و تغییرات پس از ترجمه در باقیماندهها قابل شناسایی نیستند. بنابراین، تغییرات کنفورماسیونی ناشی از تغییرات پس از ترجمه با آلفافولد فعلی قابل پیشبینی نیستند.
6) محدودیتها در روشها و تکنیکها
در نهایت باید ذکر کنیم که آلفافولد خود دارای برخی محدودیتها در روشها و تکنیکها است. به عنوان مثال: (۱) مدلهای یادگیری عمیق در حال حاضر تفسیرپذیری پایینی دارند؛ (۲) پیشبینی ساختاری آلفافولد بر اساس دادههای MSA است، یعنی تعداد زیادی توالی مرتبط با تکامل برای پیشبینی ساختارها نیاز است که ممکن است عوارض جانبی مانند سرعت پیشبینی نسبتاً کند را به همراه داشته باشد. در مقایسه، مدلهای زبانی (مانند ESMfold و RGN) امکان پیشبینی ساختار پروتئین بهصورت انتها به انتها را بهطور مستقیم از توالیهای آمینو اسید با سرعت و دقت بالا فراهم میکنند.
چالش های اخلاقی آلفافولد
استفاده از آلفافولد و فناوریهای مشابه در زمینه زیستشناسی و بیوانفورماتیک با چالشهای اخلاقی متعددی همراه است. در زیر به برخی از این چالشها اشاره میشود:
- دادههای حساس: ممکن است اطلاعات مربوط به پروتئینها و ژنها که توسط آلفافولد پیشبینی میشود، شامل دادههای حساس یا خصوصی باشد.
- استفاده نادرست از اطلاعات همانند سلاح های بیولوژیکی و میکروبی
- تغییر در روشهای تحقیق: استفاده گسترده از این فناوری ممکن است روشهای سنتی تحقیق را تحت تأثیر قرار دهد و به کاهش تنوع در رویکردهای تحقیقاتی منجر شود.
- تأثیر بر مشاغل: اتوماسیون در پیشبینی ساختار پروتئینها ممکن است به کاهش نیاز به نیروی کار انسانی در برخی زمینهها منجر شود.
خلاصه و جمع بندی از آلفافولد
عملکرد عالی آلفافولد در پیشبینی ساختار پروتئین به همراه انتشار ساختارهای بیش از 200 میلیون پروتئین پیشبینیشده توسط آن، در حال دگرگون کردن زیستشناسی ساختاری است. آلفافولد قطعاً تأثیر قابلتوجهی بر تحقیقات نیازمند اطلاعات ساختار پروتئین خواهد داشت و میتواند در بسیاری از زمینهها مانند کشف دارو، طراحی پروتئین، پیشبینی تارگت، پیشبینی عملکرد پروتئین، تعاملات پروتئین-پروتئین (PPI)، مکانیزمهای بیولوژیکی عمل و دیگر زمینهها، علاوه بر زیستشناسی ساختاری تجربی، مورد استفاده قرار گیرد. با وجود اینکه زمان بسیار کوتاهی از توسعه آلفافولد میگذرد، ما هماکنون شاهد تعدادی از کاربردهای موفق بودهایم. ما بر این باوریم که با گذشت زمان، کاربردهای بیشتری یا زمینههای جدید کاربردی توسعه خواهد یافت، به عنوان مثال، طراحی ماشینهای پروتئینی با عملکردهای پیچیده یا خاص، طراحی موجودات جدید و تشخیص بیماری. با این حال، پیشبینی آلفافولد یک درمان همهجانبه نیست و هنوز مسائل زیادی وجود دارد که نیاز به حل شدن دارند، از جمله دینامیک پروتئین، ساختارهای نواحی بینظم پروتئینها، ساختارهای جهشیافته، ساختارهای کمپلکس پروتئین-لیگاند، ساختارهای پروتئینها با تغییرات پس از ترجمه و غیره. با توسعه بیشتر الگوریتمهای هوش مصنوعی، دادههای روزافزون و قدرت محاسباتی، انتظار میرود که در آینده شگفتیهای بیشتری به ما برسد.
آلفافولد یک جهش بزرگ در حل مشکل تا شدن پروتئین است. آلفافولد تأثیر زیادی در صنعت خواهد داشت و پیشبینی تا شدن پروتئین را کمهزینهتر و در دسترستر میکند. این پتانسیل را دارد که سرعت کشفیات را در هر زمینهای که پروتئینها نقش مهمی ایفا میکنند، تسریع بخشد.
امروزه علاوه بر ساختارهای پروتئین، پیشبینی ساختار های RNA ها نیز مورد توجه است و ساختارهای پیچیده تر پروتئین و RNA ممکن است نقاط تمرکز جدید در “عصر پس از آلفافولد ” باشند.
در نهایت، ما منتظر پیشرفتهای جدید هوش مصنوعی در زیستشناسی ساختاری و دستاوردهای بیشتر در زمینه کاربرد آلفافولد در آینده هستیم.
منابع و مقالات رفرنس
https://www.nature.com/articles/s41586-021-03819-2
https://www.nature.com/articles/s42003-023-05489-4
https://www.nature.com/articles/s41392-023-01381-z
https://pubmed.ncbi.nlm.nih.gov/35757294/
https://pubmed.ncbi.nlm.nih.gov/35976160/
https://pubmed.ncbi.nlm.nih.gov/34757056/

